

What is MoLeR?
www.ai-supremacy.com

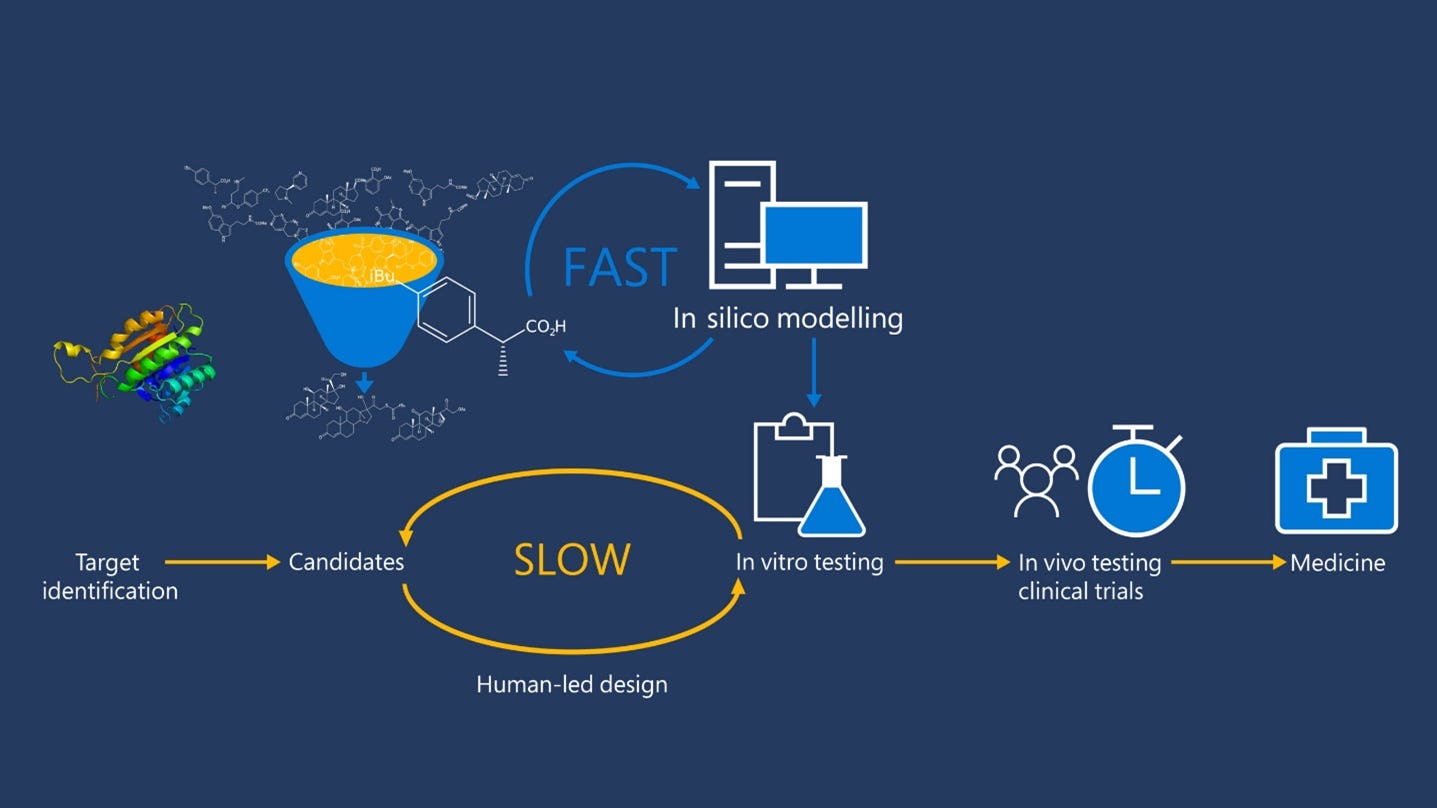
In recent years drug discovery is being more impacted by A.I. than ever before. I actually invest in this, my favorite play is $BTAI, at least if it goes down below $8 that is. I write about investing too.

We know that machine learning and other technologies are expected to make the hunt for new pharmaceuticals quicker, cheaper and more effective. Artif…
Keep reading with a 7-day free trial
Subscribe to AI Supremacy to keep reading this post and get 7 days of free access to the full post archives.